
Genesis Model Distillation
Distilling Pearl: Flow Maps for Fast All-Atom Cofolding
Modern AI models for biology have reshaped how we predict and design molecular structures. Their key capability is cofolding: simultaneously predicting the structure of a protein and any molecules bound to it, such as a drug candidate or cofactor. The dominant approach for generating all-atom biomolecular structures today centers on diffusion-based cofolding models (Ho et al., 2020), including our own state-of-the-art model, Pearl (Genesis Research Team, 2025).Pearl achieves high-fidelity structures through many small denoising steps. This accuracy comes at a proportionate inference cost, and it can be pushed further still by combining Pearl with inference-time scaling and physics- and AI-based pose ranking for challenging zero-shot predictions (see the Pearl system post).
Pearl achieves high-fidelity structures through many small denoising steps. This accuracy comes at a proportionate inference cost, and it can be pushed further still by combining Pearl with inference-time scaling and physics- and AI-based pose ranking for challenging zero-shot predictions (see the Pearl system post).
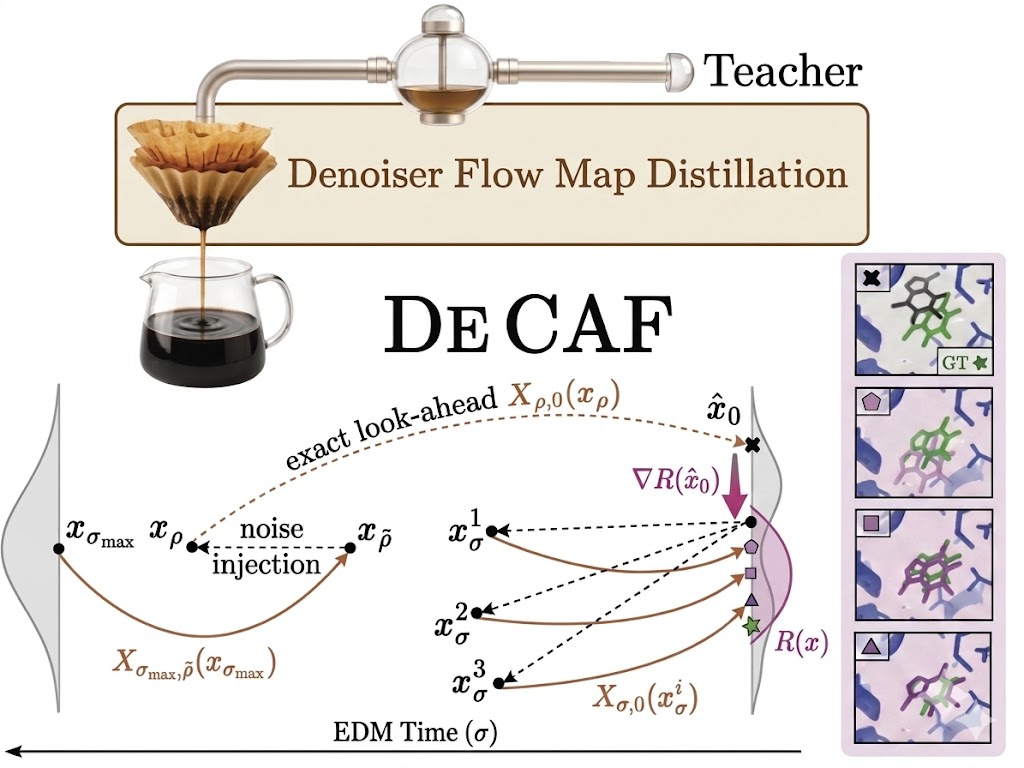
Because the molecular design space is vast, accelerating the base model offers high leverage, and this is why we developed DeCAF (Denoiser Cofolding All-atom Flowmap model) framework. DeCAF-Pearl is the first flow map model for all-atom cofolding. Instead of taking many steps along the denoising trajectory, a flow map learns to jump directly from one point on the trajectory to another, potentially traversing the entire generation process in just a handful of steps (Boffi et al., 2025).
Few-step generation unlocks two practical advantages for drug discovery and molecular design.
High-throughput virtual screening. Cofolding an entire ligand library against a target at a full diffusion-based budget is computationally challenging. A 5x inference speedup lets us screen 5x more molecules at the same compute budget.
Scalable synthetic data generation. High-quality protein-ligand complexes are the bottleneck for training downstream scoring functions, affinity predictors, and generative models. DeCAF-Pearl's 5x speedup in synthetic data generation translates directly into more training data per unit of compute, and, crucially, its success-rate parity with the teacher means this data preserves the structural signal that downstream models depend on.
Approach: How DeCAF Works
Cofolding with DeCAF-Pearl vs. Pearl-2026.1.dev. DeCAF-Pearl makes ~20x fewer model calls during diffusion sampling. Both methods also run trunk inference (~100 ms), not shown here; the full inference speedup is ~5x.
DeCAF combines three research areas: cofolding models for structure prediction (Abramson et al., 2024;Genesis Research Team, 2025), flow map distillation for speed, and inference-time steering to improve results (Singhal et al., 2025). The model learns an "average direction of travel" between any two points in time, rather than only the local direction at each instant. This "lookahead" capability is more accurate than a standard model's instantaneous prediction, which is a meaningful advantage when steering generation toward physically valid structures. Flow map models can be trained either from scratch or distilled from an existing diffusion model, making them a practical path toward faster all-atom structure generation with minimal quality gap.
Making DeCAF work required specific, non-obvious choices that we discovered only after extensive experimentation. Each decision resolves a tension that exists in all-atom biomolecular modeling but is often absent in simpler domains such as image generation. Below, we highlight the decisions that made the difference.
1. Why Time Fails: Reparameterizing to Noise-Level Space
The default move when adapting flow map methods to a new domain is to keep the time variable from the teacher. The training objective for a flow map involves a chain-rule factor relating noise level to time, and the resulting gradients are numerically unstable. Our fix is to reparameterize the entire flow map to live in σ (noise-level) space directly (Karras et al., 2022), so the problematic factor never appears.
2. Velocity vs. Alignment: Committing to Clean-Structure Prediction
The natural way to train a flow map is to match velocities — to predict the direction of motion through the trajectory. This is what Mean Flows (Geng et al., 2025) and Align Your Flow (Sabour et al., 2025) do in the image setting, and it works well there.
It works poorly for proteins. The reason is that diffusion-based cofolding models rely on a critical training trick: they predict the clean structure directly, then rigidly align the ground truth to the prediction via the Kabsch algorithm before computing the loss. This alignment accounts for the fact that a rotated or translated protein is the same protein, and it substantially reduces the variance of the gradient signal. To preserve this advantage, we parameterize DeCAF as a two-time denoiser that predicts the clean structure directly given a noisy input and two noise levels. This lets us reuse the rigid-alignment loss exactly as the teacher does.
3. DeCAF-Search: A Single Algorithm for Every Compute Budget
Prior literature on inference-time steering introduced separate methods for diffusion-based models: Boltz-1x uses Feynman-Kac steering (Singhal et al., 2025), diffusion-MCTS adapts Monte Carlo tree search (Jain et al., 2025), and our recently announced Pearl system pairs the Pearl cofolding model with novel inference-time scaling strategies.
We adapt these methods for flow maps and find that none of them is universally best. Rather than commit to one, we built DeCAF-Search as a unified algorithm that subsumes all three as special cases of a single framework: maintain a population of particles, look ahead with the flow map, refine in clean-structure space, re-noise, and reallocate compute according to a selection rule. A single implementation handles every compute regime, which lets us study which recipe wins where rather than committing to one upfront.
Results
We adopt the Pearl-2026.1.dev cofolding model, a development checkpoint of the Pearl foundation model (Genesis Research Team, 2025) trained with a 2023-12 cutoff date. We distill this foundation model directly; this effort is separate from the recently announced Pearl system which augments our production foundation model with inference-time scaling and physics+AI-based pose ranking.
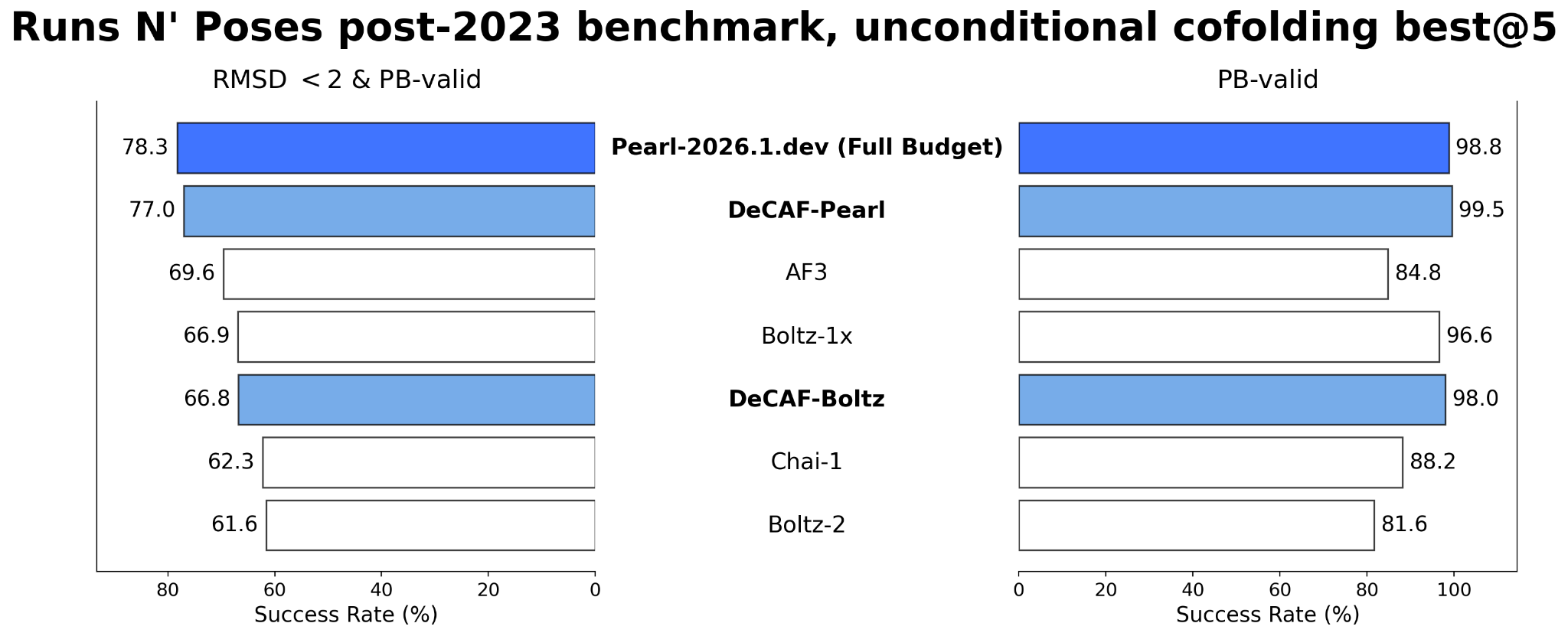
We adopt Pearl-2026.1.dev as the teacher and distill it into DeCAF-Pearl. We evaluate on the post-2023 subset of Runs N' Poses, a held-out cofolding benchmark of 196 protein-ligand structures deposited to the PDB after December 31, 2023 (Škrinjar et al., 2025). Our key findings, summarized below, confirm that DeCAF-Pearl retains most of Pearl-2026.1.dev's quality at a fraction of the compute cost, making it a practical alternative for throughput-critical applications where the quality-versus-compute trade-off is acceptable.
DeCAF-Pearl almost matches the Pearl-2026.1.dev teacher on success rate. As Figure 1 shows, against AlphaFold 3, Chai-1 Boltz-1x, and Boltz-2 — all using over 5x as many NFEs (Neural Function Evaluations) — DeCAF-Pearl wins on success rate by 7 to 15 percentage points. DeCAF-Pearl also substantially outperforms its Boltz-based counterpart (DeCAF-Boltz); as expected, a stronger base model translates into stronger DeCAF performance.
These results confirm that DeCAF-Pearl achieves state-of-the-art structure prediction quality at a fraction of the computational cost. This efficiency unlocks two key advantages: a 5x speedup over Pearl-2026.1.dev makes large-scale virtual screening tractable, enabling the rapid processing of vast ligand libraries and broadening the scope of structure-based projects. The same speedup unlocks scalable synthetic data generation, producing more high-quality protein-ligand complexes per unit of compute for training better downstream models such as scoring functions and generative models. By delivering faster inference while retaining quality, DeCAF-Pearl shortens the full cycle of molecular design.
For the full set of results, ablations, and analysis, see our arXiv preprint.
Acknowledgments
The research team at Genesis is grateful to our collaborators from Massachusetts Institute of Technology: Ron Shprints, Peter Holderrieth, Juno Nam, Rafael Gómez-Bombarelli and Tommi Jaakola; Carnegie Mellon University: Nicholas Matthew Boffi, and Joey Bose from College London and Mila.