
Zero-shot Pearl System Surpasses All Cofolding Models on OpenBind
Introduction
We would like to thank the OpenBind Consortium for their significant contribution to the field: their first public, structure-affinity benchmark for structure-based molecular AI. This carefully curated dataset is purpose-built for evaluating cofolding and docking methods against a single, well-characterized drug target and includes extensive structural coverage. Their analysis focuses on the 802 “follow-on” sets of complexes, which are non-fragment ligands with physically valid (via PoseBusters check) crystallographic poses, and their biological affinity measurements against the EV-A71 2A protease. They evaluated six cofolding models: AlphaFold3, Boltz-1, Boltz-2, OpenFold3-p2, Protenix, and RosettaFold 3, alongside several physics-based docking approaches.
We took the opportunity to evaluate our Pearl system against the full suite of published baselines. The Pearl system combines our production version of Pearl (Link), our foundation model for protein–ligand cofolding, with novel techniques for inference-time scaling along with physics- and AI-based pose ranking.
The Pearl system achieves a 78% success rate on OpenBind's joint criteria (RMSD ≤ 2 Å, PoseBusters valid, LDDT-PLI ≥ 0.8, best-of-25) in a zero-shot manner, where the Pearl system is given only a protein sequence, the ligand 2D chemical structure (SMILES), and the apo crystal structure as a template, without further knowledge of the binding site and without target-specific tuning. This surpasses all cofolding models evaluated by OpenBind by a significant margin.
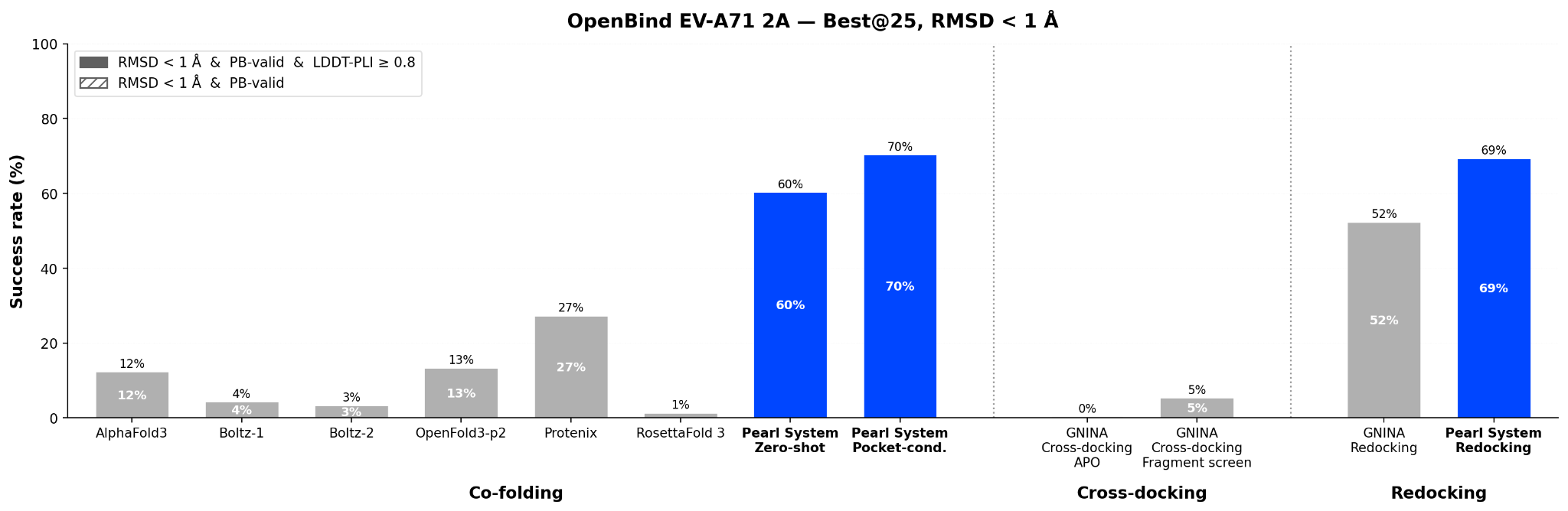
More strikingly, the zero-shot Pearl system surpasses all evaluated methods, both cofolding and redocking, at the more stringent RMSD < 1 Å threshold. We believe sub-angstrom accuracy is more relevant to real-world drug discovery, and the Pearl system achieves this on a majority of compounds.
Approach
Since the Pearl paper, we have continued to improve both the foundation model and our inference-time scaling techniques, creating the Pearl system. Specifically, our novel inference-time techniques in the Pearl system generate a diverse set of binding poses for each ligand, while using physics-based guidance to steer predictions toward chemically and geometrically valid structures. This is achieved within a comparable compute budget to standard cofolding inference. The resulting pose set is ranked by a physics- and AI-based scoring function, combining the generative capacity of a cofolding model with the discriminative power of physics- and AI-based methods.
All computational experiments use production hyperparameters without benchmark-specific tuning. This is a zero-shot setting, using the same model and inference pipeline that we deploy in active drug discovery programs. Notably, the apo template structure (PDB: 8POA) postdates Pearl's training data cutoff, confirming that the results reflect genuine generalization rather than memorization.
We evaluated three conditions:
1. Zero-shot. The Pearl system receives the protein sequence, ligand SMILES, and the apo crystal structure (PDB: 8POA) as a template, and must simultaneously predict the protein conformation, binding site location, and ligand pose.
2. Pocket-conditioned. The Pearl system additionally receives three binding-site anchor residues (S87, Q95, T126). This low resolution binding mode information is typically available in a drug discovery campaign, coming from orthogonal sources, such as: fragment screening, mutagenesis studies, or structural homology. Crucially, this information reflects realistic prior knowledge, instead of oracle information.
3. Redocking. The protein is held fixed at a crystallographic holo conformation via step-level noise-adjusted inpainting, and the Pearl system must place the ligand accordingly. This condition isolates ligand placement accuracy from protein conformation prediction, simulating a redocking procedure and facilitating comparison against “classical” docking approaches such as GNINA.
Results
Evaluation on OpenBind EV-A71 2A
Zero-shot. The Pearl system, with no privileged target-specific information, achieves a 78% success rate (RMSD ≤ 2 Å, PoseBusters valid, LDDT-PLI ≥ 0.8) and places 85% of compounds within 2 Å RMSD (PoseBusters valid), best-of-25. These metrics are substantially higher than all other cofolding models tested by OpenBind.
Pocket-conditioned. With three anchor residues from the fragment screen, the Pearl system reaches 85% on the joint criteria and 89% within 2 Å RMSD. The Pearl system achieves this feat by solely leveraging the apo crystal structure template and binding site information, without needing to fine-tune, and even surpasses fine-tuned OpenFold3-p2 (76%).
Redocking. With the holo protein fixed, the Pearl system achieves 89% on the joint criteria and 92% within 2 Å RMSD, comparable to GNINA redocking performance (85% and 92%). Note that these classic redocking methods require a ligand-bound crystal structure of the target, which is an information that is typically unavailable for novel compounds, and further assumes that the receptor conformation does not change significantly. As a reference, classical docking methods using the apo structure as evaluated by OpenBind achieve almost no success. We include GNINA cross-docking and redocking results in our figures and analyses as a reference, since GNINA was the best performing among all docking methods as evaluated by OpenBind.
This shows that the remaining gap between cofolding and redocking is small, as Pearl system’s pocket-conditioned performance (85%) already approaches its redocking ceiling (89%) and comparable to classical redocking performance.
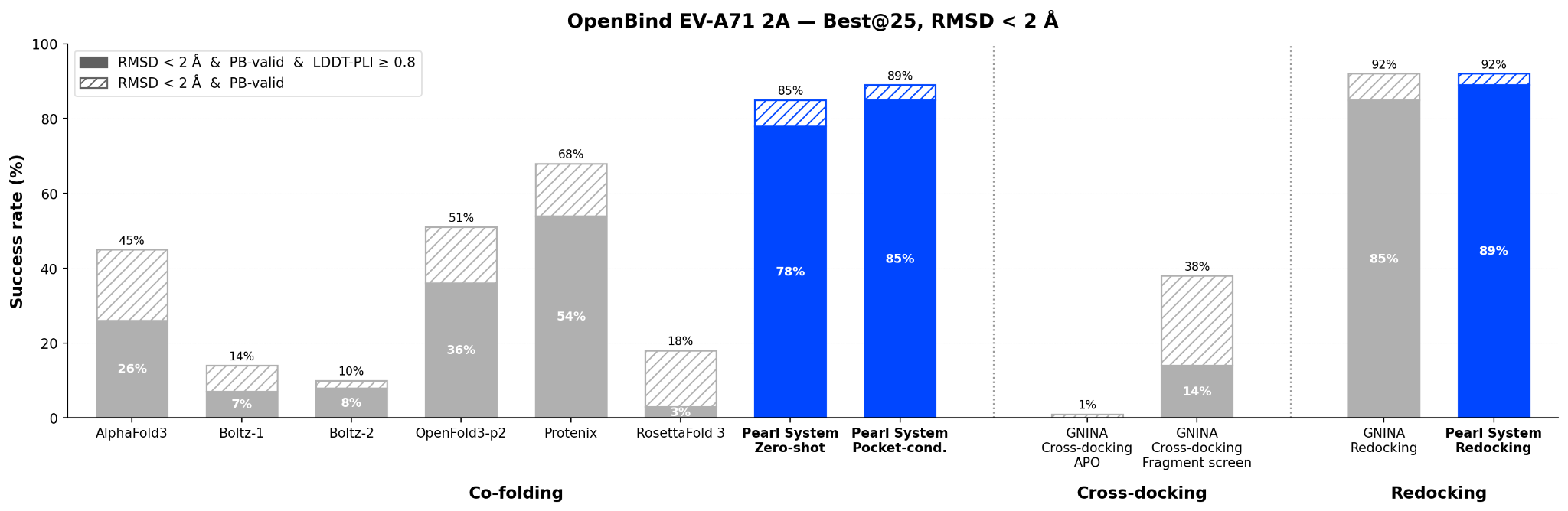
These gains are driven entirely by the Pearl system – a stronger foundation model and novel inference-time techniques – without fine-tuning on EV-A71 2A data. In practice, the Pearl system also supports ligand template conditioning from prior ligand-bound structures, a capability that is routinely used in our internal programs when “in series” cocrystal references are available.
Sub-Angstrom Accuracy
The standard RMSD < 2 Å threshold, while widely used for benchmarking, is a permissive measure of pose quality. In structure-based drug design, many medicinal chemistry strategies including: scaffold hopping, substituent optimization, and water displacement, benefit from and frequently require sub-angstrom ligand pose accuracy.
At the more demanding RMSD < 1 Å threshold, the Pearl system achieves 60% zero-shot success rate on the joint criteria and 70% with pocket conditioning, surpassing GNINA redocking performance (52%). In fact, the pocket-conditioned Pearl system matches its own redocking performance (70% vs. 69%) at this threshold, indicating that protein conformation prediction is no longer the bottleneck for sub-angstrom accuracy. For comparison, the best individual model (Protenix) only reaches 27%, success rates of other models vary between 1% to 13%.
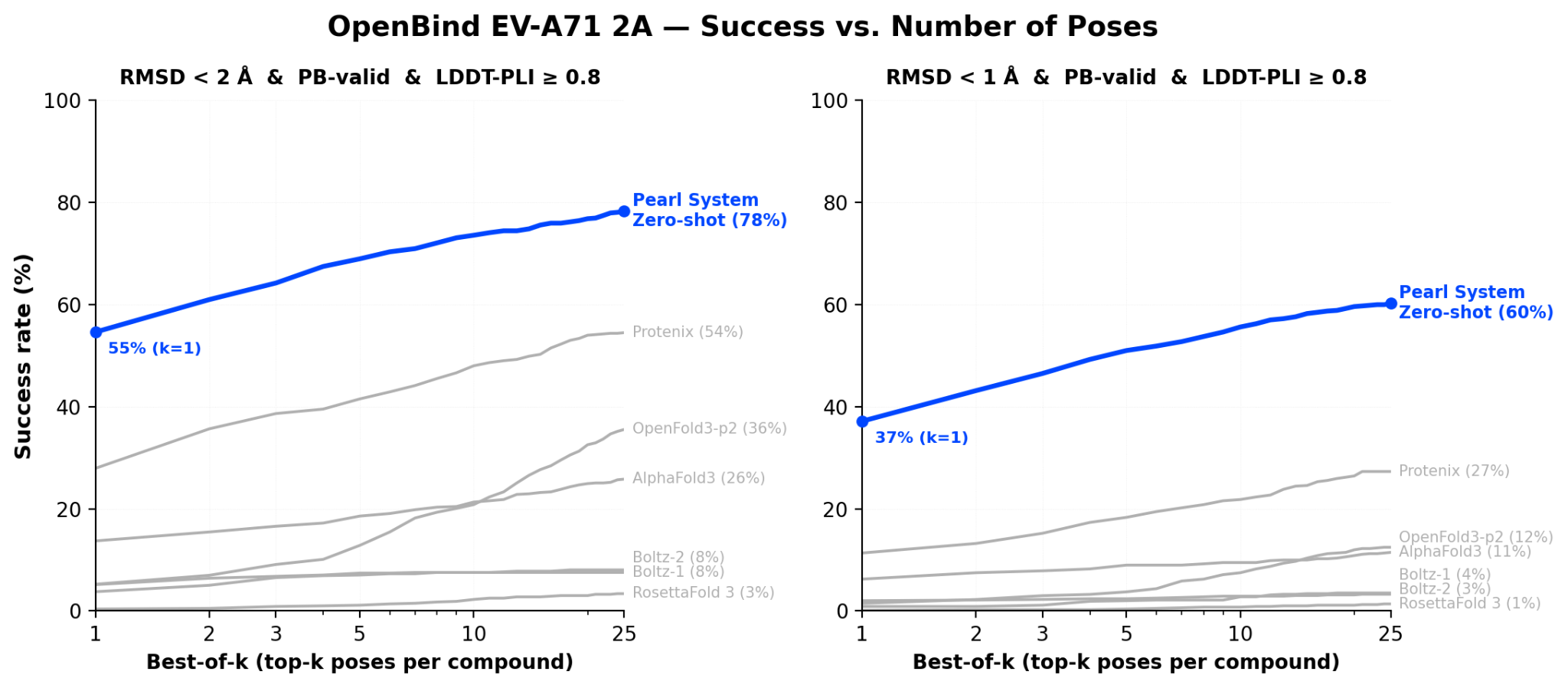
We also analyzed how the Pearl system performs across different numbers of ranked poses (best-of-k). Even with just a single pose per compound (k=1), the zero-shot Pearl system already matches or surpasses every individual cofolding model's best-of-25 pose performance, achieving 55% at RMSD < 2 Å and 37% at RMSD < 1 Å joint criteria. This further demonstrates that the gains are not just a product of generating more poses, but reflect fundamentally higher per-pose quality from the Pearl system's foundation model and inference pipeline advancements.
The Induced-Fit Challenge for EV-A71 2A Protease
EV-A71 2A protease is a challenging docking and cofolding target. A loop adjacent to the binding site (L1, residues 85–92) undergoes a large backbone rearrangement upon ligand binding, with a median Cɑ displacement of ~3.9 Å from the apo conformation. This induced-fit motion is crucial, as every one of the 802 ligand-bound structures requires this rearrangement, and the apo binding site cannot accommodate ligands without it. As such, classical docking methods using the unbound apo structure as evaluated by OpenBind only achieve < 5% success, and requires redocking with proper holo structure. The target is also underrepresented in public structural databases, meaning cofolding models cannot rely on memorized templates from training data.
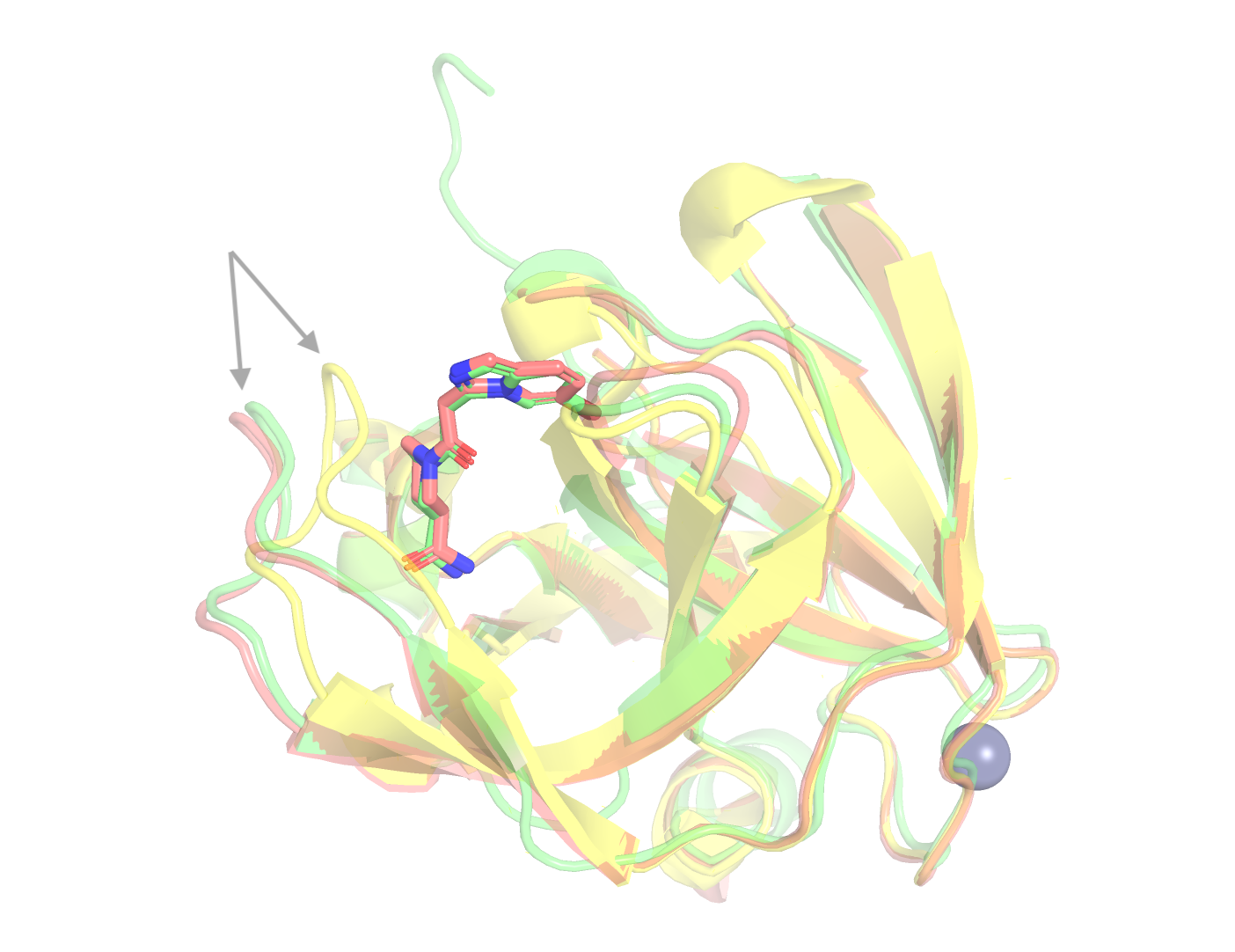
The Pearl system predicts this conformational change from sequence and ligand information alone. For example, on compound A71EV2A-x4519a, which no other zero-shot cofolding method solves, as evaluated by OpenBind, the Pearl system places the ligand within 0.28 Å RMSD of the crystallographic pose. The Pearl system correctly predicts this L1 loop rearrangement, with full PoseBusters validity and LDDT-PLI ≥ 0.8. In fact, all 25 of Pearl's top ranked poses for this compound are sub-angstrom and pass the strict quality criterion.
The capacity to model induced fit rather than assuming a rigid or pre-organized receptor is the primary driver of the performance gap between cofolding and apo docking on this target. Accounting for protein flexibility as well as achieving sub-angstrom ligand accuracy are the key reasons for Pearl’s project impact in real drug discovery campaigns.
Acknowledgments
We are grateful to our partners at NVIDIA, whose support developing Triton and cuEquivariance was instrumental in accelerating Pearl inference. We previously collaborated with NVIDIA to integrate cuEquivariance kernels and accelerate Pearl achieving 10-80% speedup for inference. Migrating from H100 to B200, Pearl achieved a 1.65x inference speed boost. With further software optimizations, Pearl achieved a total 2.3–2.8x end-to-end speedup on B200 vs baseline H100 code. The throughput gains we have unlocked with NVIDIA make it practical to run inference at industrial scale as well as generate high-quality synthetic data and train substantially larger models. We thank the NVIDIA team for their technical guidance and collaboration throughout this effort.
We thank the OpenBind Consortium for this benchmark, the crystallographic data, and the thorough analysis of baselines and failure modes. Public benchmarks with well-characterized baselines are essential for driving progress in computational structure-based drug design, and OpenBind is providing a valuable contribution to the community.